
วอนภาครัฐอย่าทิ้งผู้ป่วยไว้ข้างหลัง…เร่งผลักดันการเข้าถึงการรักษาโรคหายากโรคโกเชร์ เป็นหนึ่งในตัวอย่างของ โรคหายาก ที่มีกว่า 5,000 – 6,000 โรค และเป็นหนึ่งในตัวอย่างโรคหายากในกลุ่มแอลเอสดีซึ่งมีมากว่า 50 โรค โดยโรคโกเชร์เป็นเพียงโรคในกลุ่มแอลเอสดีโรคเดียวที่ผู้ป่วยเข้าถึงการรักษาในประเทศไทย เนื่องจากยาได้รับการบรรจุอยู่ในบัญชียาหลักแห่งชาติ ตั้งแต่ปี 2556 จากแรงขับเคลื่อนของภาคส่วนต่างๆ ทั้งภาครัฐที่เห็นความสำคัญของการเข้าถึงการรักษาและพิจารณาอนุมัติงบประมาณอย่างต่อเนื่อง ตลอดจน แพทย์ผู้เชี่ยวชาญ และมูลนิธิผู้ป่วย ทำให้การเข้าถึงการรักษาโรคโกเชร์ ถือเป็นโมเดลแห่งความหวังของผู้ป่วยโรคหายากอื่นๆ ที่ยังไม่ได้รับสิทธิและยังคงรอโอกาสเปลี่ยนแปลงของสังคมไทย เพื่อเข้าถึงการรักษาที่มีประสิทธิภาพอย่างปลอดภัยและยั่งยืน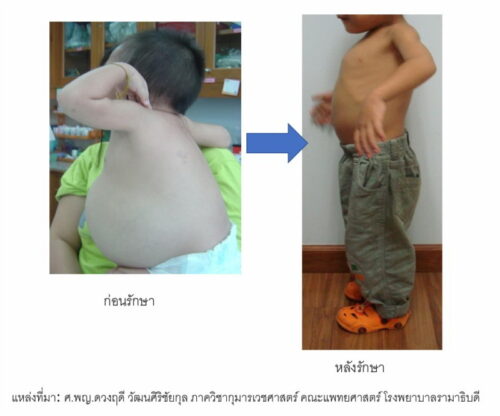 เนื่องในวันโรคโกเชร์สากล (International Gaucher Day) ซึ่งตรงกับทุกวันที่ 1 ตุลาคมของทุกปี ศ.พญ. ดวงฤดี วัฒนศิริชัยกุล แพทย์ผู้เชี่ยวชาญสาขาเวชพันธุศาสตร์ ภาควิชากุมารเวชศาสตร์ โรงพยาบาลรามาธิบดี มหาวิทยาลัยมหิดล จึงได้เผยแพร่ข้อมูลเกี่ยวกับโรคโกเชร์ เพื่อกระตุ้นให้เกิดการรับรู้ เกี่ยวกับโรคโกเชร์ รวมถึงโรคหายากอื่นๆ ในวงกว้าง เพื่อร่วมกันสร้างสังคมคุณภาพโดยไม่ทิ้งใครไว้ข้างหลัง
เนื่องในวันโรคโกเชร์สากล (International Gaucher Day) ซึ่งตรงกับทุกวันที่ 1 ตุลาคมของทุกปี ศ.พญ. ดวงฤดี วัฒนศิริชัยกุล แพทย์ผู้เชี่ยวชาญสาขาเวชพันธุศาสตร์ ภาควิชากุมารเวชศาสตร์ โรงพยาบาลรามาธิบดี มหาวิทยาลัยมหิดล จึงได้เผยแพร่ข้อมูลเกี่ยวกับโรคโกเชร์ เพื่อกระตุ้นให้เกิดการรับรู้ เกี่ยวกับโรคโกเชร์ รวมถึงโรคหายากอื่นๆ ในวงกว้าง เพื่อร่วมกันสร้างสังคมคุณภาพโดยไม่ทิ้งใครไว้ข้างหลัง
โรคโกเชร์ (Gaucher Disease) เกิดจากความผิดปกติทางพันธุกรรม โดยส่วนใหญ่มักแสดงอาการตั้งแต่เด็ก หรือในบางคนอาจแสดงอาการตอนเป็นผู้ใหญ่ โรคโกเชร์ แบ่งออกเป็น 3 ชนิด อาการแสดงของโรคโกเชร์รวมถึง ตับม้ามโต อาการทางกระดูก และระบบประสาท ส่วนใหญ่คนไทยมักจะเป็นโกเชร์ชนิดที่ 1 และชนิดที่ 3 โดยแรกคลอดจะดูปกติ แต่พออายุ 1-2 ขวบ อาจสังเกตได้ว่าท้องจะมีขนาดใหญ่ขึ้น เนื่องจากตับและม้ามโต พบภาวะซีด เกล็ดเลือดต่ำ โดยทั่วไปแพทย์อาจจะนึกถึงโรคธาลัสซีเมียซึ่งเป็นโรคที่มีอาการใกล้เคียงกันและคนไทยเป็นมากกว่าทำให้ไม่ได้นึกถึงโรคโกเชร์ นอกจากนี้โรคโกเชร์ที่แสดงอาการในเด็ก ส่วนใหญ่เป็นชนิดที่ 3 มีอาการรุนแรงกว่าโรคโกเชร์ที่แสดงอาการในผู้ใหญ่ จึงขอฝากถึงแพทย์ทั่วไปว่าหากตรวจแล้วพบอาการโรคที่ไม่ตรงไปตรงมา ให้สงสัยได้ว่าคนไข้อาจเป็นโรคหายากโรคใดโรคหนึ่ง หรือโรคโกเชร์ ควรส่งต่อให้แพทย์ด้านพันธุกรรมหรือแพทย์โรคเลือด เพื่อนำเข้าสู่กระบวนการวินิจฉัยและรักษาต่อไป
ปัจจุบันสามารถตรวจยืนยันว่าเป็นโรคโกเชร์ ได้ถึง 3 วิธีคือ 1) นำเลือดมาตรวจเอนไซม์เม็ดเลือดขาว โดยโรงพยาบาลรามาธิบดี สามารถตรวจได้ภายใน 2-3 วันก็ทราบผล 2) ตรวจสารในเลือด หรือตรวจ Lyso-Gb1 เป็นสารที่บ่งชี้ว่าเป็นโรคโกเชร์ ซึ่งผู้ป่วยโกเชร์จะมีสารชนิดนี้สะสมอยู่ในเลือด และ 3) ตรวจความผิดปกติของยีนเพื่อยืนยันโรคโกเชร์ ทั้งนี้ หากตรวจพบ 1 ใน 3 หรือ 2 ใน 3 วิธีนี้ก็สามารถยืนยันได้ว่าเป็นโรคโกเชร์ ปัจจุบันแพทย์จะแนะนำให้ครอบครัวที่มีประวัติหรือมีญาติที่ป่วยเป็นโกเชร์ เข้ารับการตรวจเพราะเป็นกลุ่มเสี่ยง เนื่องจากโรคโกเชร์เกิดจากพันธุกรรมจากความผิดปกติของยีนซึ่งมักพบยีนแฝง (หรือพาหะ) ในทั้งพ่อและแม่ จากงานวิจัยในคนไทยตรวจพบคนเป็นพาหะโรคโกเชร์ 1 คน จาก 200 คน หรือคิดเป็น 0.5% วิธีการรักษาโรคโกเชร์ มี 3 วิธี ได้แก่
1. ยาเอ็นไซม์ทดแทน โดยการให้ยาเข้าทางหลอดเลือดดำทุก 2 สัปดาห์ เพราะยานี้จะทำงานได้ดีประมาณ 2 สัปดาห์ จากผู้ป่วยที่ท้องป่องเหมือนลูกโป่ง พอรักษาไปสักพัก ท้องยุบกลับมาเป็นปกติ ตับม้ามยุบหาย หายจากอาการซีดและเกล็ดเลือดกลับมาเป็นปกติ เด็กโตขึ้นและมีพัฒนาการเช่นเด็กปกติ วิธีนี้จำเป็นต้องได้รับยารักษาอย่างต่อเนื่องไปตลอดชีวิต
2. ยากิน อาจจะยังไม่เหมาะกับผู้ป่วยคนไทยและคนเอเชียเนื่องจากส่วนใหญ่จะเป็นโกเชร์ชนิดที่มีอาการค่อนข้างรุนแรง การรักษาแบบยากินอาจเหมาะกับชาติตะวันตกที่มีอาการไม่รุนแรงมากเท่าผู้ป่วยไทย
3. การปลูกถ่ายสเต็มเซลล์เม็ดเลือด เป็นวิธีที่หายขาดจากโรคได้ แต่ก็มีความเสี่ยง แพทย์จำเป็นต้องพูดคุยกับครอบครัว กรณีที่ปลูกถ่ายไขกระดูกแล้ว เม็ดเลือดสเต็มเซลล์ติด ก็จะต้องให้ยากดภูมิในช่วง 2 ปีแรก หลังจากนั้นคือหายขาด แต่กระบวนการที่จะหาไขกระดูกที่เข้ากันก็ไม่ง่าย ส่วนกระบวนการปลูกถ่ายไขกระดูก ก็มีความเสี่ยง โดยอัตราการเสียชีวิตอยู่ที่ 8%โดยส่วนใหญ่ในประเทศไทย แพทย์จะแนะนำการรักษาแบบผสมผสาน โดยในช่วงแรกที่ร่างกายยังอ่อนแอ แนะนำให้ยาเอ็นไซม์ทดแทนไปก่อนประมาณ 2-3 ปี จนสภาพร่างกายดีขึ้นกลับมาเป็นปกติ ระหว่างนี้ก็หาสเต็มเซลล์ที่เข้ากันได้ไปก่อนแล้วค่อยปลูกถ่ายไขกระดูกในภายหลัง ในฐานะผู้ปกครองผู้ป่วยโรคโกเชร์ นายบุญ พุฒิพงศ์ธนโชติ ประธานมูลนิธิโรคพันธุกรรมแอลเอสดี เล่าว่าย้อนไปเมื่อ 17-18 ปีที่แล้ว ตอนนั้นลูกอายุ 2 ขวบกว่ามีไข้ขึ้นสูงจึงพาไปโรงพยาบาล โชคดีที่หมอตรวจร่างกายละเอียดและคลำเจอตับม้ามโตผิดปกติ จึงเกิดความสงสัยว่าจะเป็นโรคทางพันธุกรรม ซึ่งก็ใช้เวลาตรวจนานถึง 2 เดือน จนทราบว่าลูกป่วยเป็นโรคโกเชร์ ที่น่าตกใจคือค่ายารักษาโรคนี้สูงถึง 2 ล้านบาทต่อปี และต้องรักษาไปตลอดชีวิต ช่วงแรกเหมือนชีวิตสิ้นหวังคิดวนอยู่ในหัวว่าควรทำอย่างไร และได้ปรึกษาคุณหมอว่าจะรวบรวมคนไข้ที่ป่วยเป็นโรคหายากที่เกิดจากพันธุกรรมแอลเอสดี ซึ่งนอกจากโรคโกเชร์แล้ว ยังมีอีกหลายโรคมาร่วมพูดคุยกันเพื่อหาทางออก จึงเป็นที่มาของการก่อตั้งมูลนิธิโรคพันธุกรรมแอลเอสดี มีการจัดตั้งคณะทำงานประกอบด้วยแพทย์ด้านพันธุกรรมและกลุ่มผู้ปกครองหลังจากที่ก่อตั้งมูลนิธิฯ คณะทำงานได้รวบรวมสถิติข้อมูลต่างๆ และได้นำเสนอเรื่องไปยังบัญชียาหลักแห่งชาติเพื่อให้พิจารณาบรรจุยารักษาโรคโกเชร์ เนื่องด้วยโรคโกเชร์สามารถรักษาหายขาดได้และไม่เป็นภาระงบประมาณมากนัก โดยในปี 2556 ยาได้ถูกบรรจุเข้าบัญชียาหลักแห่งชาติ ทำให้ผู้ป่วยสิทธิบัตรทอง สิทธิประกันสังคม หรือสิทธิสวัสดิการรักษาพยาบาลข้าราชการ สามารถเข้าถึงการรักษาและเบิกยาได้ โดยในปีแรกสามารถรักษาผู้ป่วยโกเชร์ได้ถึง 12 คน และปัจจุบันมีผู้ป่วยที่ได้รับการรักษาทั้งสิ้น 23 คน และเคยมีผู้ป่วยโรคโกเชร์ที่ได้รับการปลูกถ่ายไขกระดูกและหายขาดแล้ว 6 คน อย่างไรก็ดี ยังมีข้อจำกัดในเรื่องการหาไขกระดูกที่เข้ากันได้ ซึ่งไม่ใช่เรื่องง่าย เพราะหากเข้ากันไม่ได้ร่างกายจะสร้างปฎิกิริยาต่อต้านและอาจเป็นอันตรายอย่างรุนแรงถึงแก่ชีวิตได้
ศ.พญ. ดวงฤดี กล่าวเสริมว่า โรคหายากในแต่ละโรค จะมียารักษาที่ราคาแตกต่างกันไป บางโรคค่ายารักษา 2-3 แสนต่อปี บางโรคหลักล้านต่อปี ซึ่งปัจจุบันมีเทคโนโลยีและวิธีการรักษาใหม่ๆ เช่น ยีนบำบัด (Gene Therapy) โดยจะมีการปรับแต่งยีนที่ผิดปกติใส่เข้าไปในคนไข้เพื่อให้หายจากโรค รักษาเพียงครั้งเดียวแต่หายตลอดชีวิต ค่ารักษาประมาณ 50–60 ล้านบาท บางคนอาจมองว่าราคาสูงมาก แต่หากเทียบกับค่ารักษาปีละ 2 ล้านบาทต่อคน หากรักษายาวนาน 50 ปี ก็เท่ากับ 100 ล้านบาท เพราะโรคหายากจะต้องรักษาไปตลอดชีวิต ทำให้ต้องใช้งบประมาณมากกว่าหลายเท่า จึงทำให้มีการคิดวิธีรักษาแบบใหม่ คือ ‘ยีนบำบัด’ ฉีดครั้งเดียวแต่หายขาด คุณภาพชีวิตก็ดีขึ้นมาก ครอบครัวและผู้ป่วยไม่ต้องทนทุกข์ทรมาน กลับมาใช้ชีวิตปกติและเป็นทรัพยากรที่สำคัญของชาติต่อไปได้แม้ว่าปัจจุบัน โรคโกเชร์ยังไม่มีการรักษาด้วยยีนบำบัด แต่โรคหายากบางโรคมีการรักษาด้วยวิธีนี้แล้ว จึงจำเป็นต้องให้สังคมไทยรับรู้เพื่อให้คนไข้มีโอกาสเข้าถึงการวินิจฉัยได้รับการรักษาก่อนที่จะเกิดภาวะแทรกซ้อนรุนแรง ซึ่งหากเกิดภาวะแทรกซ้อนรุนแรงแล้วการได้รับยาก็ช่วยอะไรไม่ได้มาก การสร้างการรับรู้ให้กับสังคมเพื่อสร้างแรงขับเคลื่อนไปสู่ภาครัฐจึงเป็นสิ่งจำเป็น ทั้งนี้ ศ.พญ. ดวงฤดี วอนให้ฝ่ายที่เกี่ยวข้องกับนโยบายการเงิน สิทธิประโยชน์ทั้งบัตรทอง บัตรราชการ ประกันสังคม รวมไปถึงสำนักงานคณะกรรมการอาหารและยา (อย.) ร่วมกันพิจารณาหารือถึงกระบวนการ และแนวทางใหม่ๆ ที่เหมาะสมกับการเข้าถึงรักษาโรคหายาก ถึงงบประมาณจะสูง แต่จำนวนผู้ป่วยมีน้อยราย ผลกระทบงบประมาณไม่สูงมาก อีกประการหนึ่ง โรคหายากจำเป็นต้องได้รับการรักษาไม่เช่นนั้นอาจเป็นอันตรายถึงชีวิตก่อนวัยอันควร อีกความท้าทายสำคัญ คือ จำนวนแพทย์ด้านพันธุกรรม ซึ่งยังมีน้อยอยู่ ทั้งประเทศไทยมีเพียง 28 คน อยู่ภาคเหนือ 1 คน ภาคตะวันออกเฉียงเหนือ 1 คน และนอกนั้นอยู่ในกรุงเทพฯ ด้วยปัญหาการเบิกจ่ายไม่ค่อยได้ เช่น ค่าตรวจยีนเบิกไม่ได้ ค่ายารักษาก็เบิกจ่ายไม่ได้ จึงไม่มีใครอยากเรียนสาขานี้ เพราะแพทย์ก็อยากให้คนไข้ได้เข้าถึงการรักษาที่เหมาะสม ซึ่งต้องยอมรับว่าทุกวันนี้ยังมีผู้ป่วยโรคหายากอีกมากที่เข้าไม่ถึงระบบสุขภาพของสาธารณสุขไทย
ศ.พญ. ดวงฤดี ฝากไปยังทุกภาคส่วนที่เกี่ยวข้องว่า ทุกวันนี้ โรคหายากหรือโรคทางพันธุกรรมสามารถรักษาได้ และมีกรอบทิศทางที่ดำเนินการได้ การคัดกรองโรคจะทำให้ตรวจวินิจฉัยได้รวดเร็วขึ้น ฉะนั้น จึงวอนขอให้ภาครัฐพิจารณาส่งเสริมการผลิตแพทย์ผู้เชี่ยวชาญด้านพันธุกรรมเพิ่มเติม เพื่อบรรจุและส่งไปประจำยังเขตสุขภาพ 13 เขตทั่วประเทศ เพื่อให้การวินิจฉัยเป็นไปอย่างถูกต้องและรวดเร็ว เพราะยิ่งตรวจพบเร็วเท่าไร คนไข้ก็สามารถเข้าถึงการรักษาเร็วเท่านั้น และยังเป็นประโยชน์ป้องกันโรคเกิดซ้ำอีกในครอบครัวซึ่งนับว่าคุ้มค่าอย่างยิ่งแก่ครอบครัว นอกจากนี้ ยังเสนอให้ภาครัฐพิจารณาปรับนโยบายเกี่ยวกับกระบวนการพิจารณายา และงบประมาณ ให้เข้ากับบริบทของสังคมและสอดคล้องกับนวัตกรรมทางการแพทย์ เพื่อเร่งผลักดันการเข้าถึงการรักษาโรคหายากที่มีประสิทธิภาพ ปลอดภัยและยั่งยืน




More Stories
MEDEZE เปิดบ้านรับ THAI VI โชว์ศักยภาพธุรกิจสเต็มเซลล์ มองโอกาสเติบโตเวชศาสตร์ฟื้นฟูหนุนเศรษฐกิจสุขภาพ
MEDEZE จับมือ จุฬาลงกรณ์มหาวิทยาลัย ประกาศความสำเร็จของโครงการ สู่การส่งมอบเทคโนโลยีการผลิตโปรตีนรีคอมบิแนนท์
บำรุงราษฎร์ภูเก็ต ร่วมงาน Phuket Health Expo 2026 ตอกย้ำเครือข่ายการดูแลสุขภาพเพื่อชาวภูเก็ตและอันดามัน